Некоторые особенности гипербилирубинемии.

Билирубин крови представляет из себя гидрофобную молекулу, полученную путем последовательной деградации гема (комплексного соединения порфирина и железа 2+) при расщеплении гемсодержащих белков: гемоглобина, миоглобина и системы цитохромов. Схема метаболизма выглядит достаточно просто: гем -> биливердин -> билирубин. Некоторым биохимикам важно понимать, что биливердин -> билирубин это реакция восстановления. Поэтому при некоторых условиях (о них в примечании) билирубин с «удовольствием» окислится назад в биливердин.
Поучившийся билирубин – малорастворимое в воде вещество и в роли «транспорта» для него выступает молекула альбумина. Именно связка билирубин-альбумин и присутствует в крови, как СВОБОДНЫЙ (неконъюгированный, непрямой) билирубин. Свободный билирубин, концентрации которого превосходят связывающие способности альбумина – достаточно токсическое для человека вещество, проникающее и в клетки, и через гематоэнцефалический барьер. Последнее может привести к ядерной желтухе у новорожденных и серьезной нейротоксичности с поражением слуха, параличам, апноэ и тд. Физиологически важно помнить: связывающая способность альбумина падает при нарастании ацидоза и повышении температуры.
Коньюгация свободного билирубина из крови происходит в эндоплазматическом ретикулуме гепатоцитов, в микросомах. Вот тут и начинаются некоторые интересные подробности этого процесса. Первое, и это важно для дальнейшего понимания, гепатоцит имеет 2 «полюса» : базальная или синусоидальная часть, которая обращена в кровоток. И апикальная часть, которая обращена в желчеток. Т.е. – это некий небольшой цех, куда с одной стороны (через базальную часть) заходит неконъюгированный (свободный) билирубин, в цеху происходит его обработка (конъюгация) и продукт ( конъюгированный билирубин) выходит с другой стороны (через апикальную часть).
Захват и транспортировка билирубина из крови осуществляется посредством ворсинок базальной (синусоидальной) части и благодаря ферменту глутатион-S-трансферазе. Именно этот фермент транспортирует неконъюгированный билирубин к микросомам гепатоцита, где и происходит конъюгация.
Захват «танцующего» свободного билирубина из крови осуществляется Глутатион-S-трансферазой.
Таким образом, захваченный свободный билирубин доставляется этим ферментом к микросомам гепатоцитов, где и начинается его конъюгация с обязательным участием фермента UDP-glucuronyl-transferase. (Уридин – 5-ДиФосфат глюкуронилтрансфераза = УДФ-глюкуронилтрансфераза = УДФ-ГТ)
Превращение неконъюгированного билирубина (свободного) в конъюгированный (связанный с глюкуроновой кислотой) происходит в микросомах гепатоцитов с обязательным участием УДФ-глюкуронилтрансферазы.
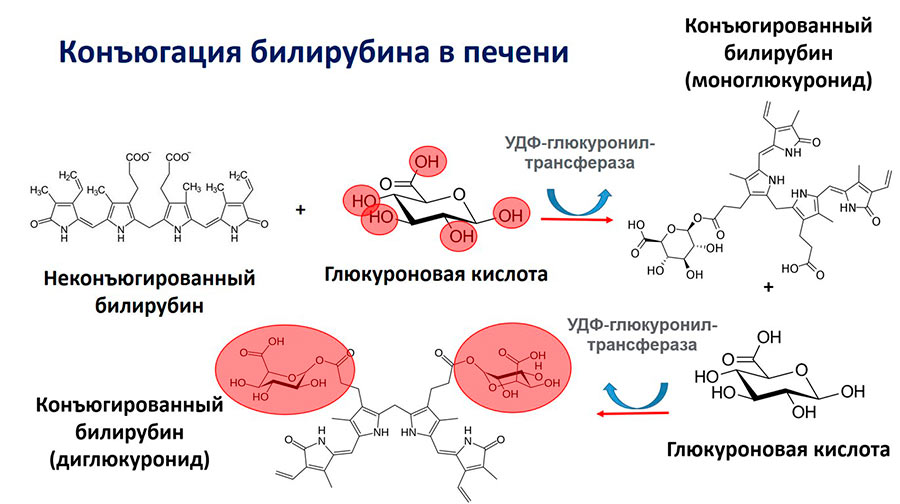
Получающийся конъюгированный билирубин бывает в 2 формах: моно и диглюкуронид, но это не особо важно. И та, и другая форма конъюгированного билирубина должны покинуть гепатоцит и выйти в желчевыводящие синусы и каналы. В норме это происходит через апикальную часть гепатоцита, граничащую с желчеотводящим синусом. Но разумно задать вопрос: свободный билирубин транспортируется в «цех» конъюгации Глутатион-S-трансферазой, там он конъюгируется в присутствии УДФ-глюкуронилтрансферазы. И выбрасывается в цитоплазму. Что его заставляет двигаться или направляет к апикальной, желчеотводящей части гепатоцита? А ни что… Конъюгированный билирубин просто накапливатся в гепатоците, создавая градиент концентрации. Но позвольте – он так же тогда может выйти в кровоток через базальную, захватывающую часть. Да, может. Но есть 2 нюанса.
Дело в том, что на базальной мембране находятся 2 хитрых полипептида: OATP1B1 и OATP1B3. Они обеспечивают ОБРАТНЫЙ захват конъюгированного билирубина, не давая ему проникнуть в кровь. И в норме все, что выбрасывает синусоидальная помпа ABCC3 базальной мембаны гепатоцита в кровоток они захватывают назад.
А вот на апикальной части стоят вполне себе работающие и предназначенные для этого «насосы», откачивающие конъюгированный билирубин в желчеток : это канальцы апикальной части гепатоцита, которые контролирует белок MRP2 (Multidrug resistance-associated protein 2), обеспечивающий транспорт органических анионов и кодируемый геном ABCC2.
Т.е. мы получаем вполне себе работающую систему отвода конъюгированного билирубина по одному вектору: микросомы -> апикальная часть. Вместе с тем понятно, что нарушиться или поломаться вся эта структура… как говорят в Одессе: имеет место где.
Вывод образующегося конъюгированного билирубина осуществляется через открытые канальцы апикальной части гепатоцита, которые контролируются белком MRP2. Обратный проход в кровоток через базальную часть закрыт специализированными белками OATP1B1 и OATP1B3.
Чаще всего нарушения процесса конъюгации могут быть на 3 уровнях:
- В силу некоторых причин связка билирубин-альбумин ПЛОХО захватывается ворсинками базальной части гепатоцита. Это – патология захвата (фермент Глутатион-S-трансфераза), конечно будет расти СВОБОДНЫЙ (неконъюгированный билирубин)
- В силу генетических особенностей неполноценно работает фермент участием УДФ-глюкуронилтрансфераза. Это приводит к нарушениям процесса конъюгации в той или иной степени, свободный билирубин стоит в очереди на обработку, а его все не зовут и не зовут. Количество свободного билирубина растет. Это – патология конъюгации.
- Вывод прямого (конъюгированного) билирубина осуществляются через канальцы апикальной части гепатоцита. Генетическая патология MRP2 каналикулярного белка приводит к тому, что уже конъюгированный гемоглобин накапливается в гепатоците и через задние (базальные) ворота все же начинает проникать в кровь. (белки OATP1B1 и OATP1B3 просто не справляются). В крови растет конъюгированный билирубин. Это – патология вывода.
Далее - основные синдромы и патофизиология процессов, которые связаны с нарушением процесса конъюгации билирубина.
СИНДРОМ ЖИЛЬБЕРА (Gilbert syndrome)
Эта- доброкачественная свободная билирубинемия, хронически протекающая болезнь. Была впервые диагностирована в 1901 году французским гастроэнтерологом Огюстеном Николя Жильбером. В патогенезе синдрома лежит нарушение захвата билирубина из-за нарушения его транспорта глутатион-S-трансферазой, доставляющей неконъюгированный билирубин к микросомам гепатоцитов, а также неполноценность фермента микросом уридиндифосфатглюкуронилтрансферазы (той самой УДФ-глюкуронилтрансферазы), при помощи которого осуществляется конъюгация билирубина с глюкуроновой кислотой.
Фактически мы имеем и патологию захвата ( из-за транспорта), и медленное конъюгирование.
Внимание – не патологическое конъюгирование, а медленное. УДФ-глюкуронилтрансфераза работает! Но не так активно, как хотелось бы. Падение активности обычно не превышает 30% от нормы. Однако в реальности на выраженность симптомов и выраженность свободной (непрямой билирубинемии) основное действие оказывает не транспорт, а именно неполноценная активность УДФ-глюкуронилтрансферазы. Но все же помним, что и транспорт может быть при чем, если он – единственное место нарушения - ну тогда это уж очень «мягкий» Жильбер будет.
Основная причина синдрома Жильбера – падение активности катализатора конъюгации – фермента УДФ-глюкуронилтрансферазы. Активность нарушена обычно в диапазоне до 30%.
Морфологическое повреждение обычно характеризуется умеренной жировой дистрофией печени и то не всегда. Уровень билирубина редко превышает 100 мкмоль/л и то – это бывает во время провоцирующих событий: инфекция, голодание, стресс, физическая нагрузка. Диагноз синдрома Жильбера выставляется как первая линия сразу, если нет данных в пользу повреждения печени (АСТ-АЛТ), и обструкционных проблем (ГГТ-ЩФ) и есть данные по флуктуации свободного билирубина крови в пределах 30-70 мкмоль/л.
Лечение обычно не требуется. У мужчин встречается чаще чем у женщин от 2 до 7 раз. Примерно от 3 до 7 % популяции западных стран поражены с-ом Жильбера. На что стоит обратить внимание – так это проверить нет ли усиленного (скрытого) гемолиза, что так же приводит к свободной гипербилирубинемие: анализ крови на морфологию эритроцитов, ретикулоциты, гаптоглобин. Учитывая, что может быть свободная гипербилирубинемия от недостаточной активности УДФ-глюкуронилтрансферазы и + усиленного гемолиза некоторые авторы выделяют:
- синдром Жильбера 1 - недостаточность только УДФ-ГТ;
- синдром Жильбера 2 -недостаточность УДФ-ГТ и сопутствующий гемолиз .
СИНДРОМ КРИГЛЕРА — НАЙЯРА (Crigler Najjar syndrome)
А вот это уже злокачественная свободная билирубинемия. Но она характерна для новорожденных, характеризуется ПОЛНОЙ или ЗНАЧИТЕЛЬНОЙ неактивностью УДФ-глюкуронилтрансферазы, что приводит к резкому росту свободной билирубинемии до цифр 200-400 мкмоль /л и выше. Бывает I и II типов, особенно злокачественная – I типа, когда практически полное отсутствие данного фермента и в желчи просто нет прямого билирубина. Такие пациенты редко доживают до 2 лет, обычно смерть наступает в течение первых суток/месяцев жизни из-за повреждения нервной системы: токсическая энцефалопатия (повреждаются клетки базальных ядер головного мозга), что приводит к системной органной недостаточности. Очень редкая патология.
II тип более «мягкий», флуктуации билирубина происходят на уровнях 100-430 мкмоль/л, в крови определяется и небольшое количество прямого билирубина ( конъюгация очень слабо, но идет), неплохой результат дает применение фенобарбитала (стимулирует УДФ-глюкуронилтрансферазу) и фототерапия – разрушает свободный билирубин в тканях. Встречается чаще.
Основная причина синдрома Криглера - Найяра – полное отсутствие или существенное падение активности катализатора конъюгации – фермента УДФ-глюкуронилтрансферазы.
Билирубин поглощает синий свет с длиной волны 450—460 нм, при этом происходят процессы поворота его колец вокруг центральной оси молекулы (изомеризация) , котрые трансформируют билирубин в малотоксичный и выводящийся люмирубин (вернее люмирубины, там несколько видов).
Следующие 2 поражения, характеризуются прямой (конъюгированной) гипербилирубинемией. Соответственно захват билирубина из крови нормальный, конъюгация нормальная, а потом что-то идет не так. Что?
Синдром Дабина – Джонсона (Dubin-Johnson syndrome)
генетический дефект каналикулярного белка MRP2, который выводит конъюгированный билирубин через апикальную часть гепатоцита в желчные протоки. Конъюгированный гемоглобин накапливается в гепатоцитах – что вызывает регургитацию его в кровоток и извращение метаболизма адреналина, приводящее к накоплению меланина в печени – шоколадная печень. В 70 % случаев синдром Дабина — Джонсона проявляется в молодом возрасте (до 20 лет), очень редко у людей старше 50 лет.
Заболевание очень редкое, не влияет на продолжительность жизни пациентов. Но клиническая симптоматика обычно более выражена, чем при других мягких поражениях: повышенная утомляемость, плохой аппетит, боли в правом подреберье, диарея. Желтуха может быть постоянной, а также сопровождаться нерезким кожным зудом. Диспепсические кризы, самочувствие всегда плохое. У некоторых больных заболевание десятилетиями протекает бессимптомно.
Основная причина синдрома Дабина – Джонсона – генетический дефект каналикулярного белка MRP2, который выводит конъюгированный билирубин через апикальную часть гепатоцита в желчные протоки.
Синдром Ротора (Rotor syndrome)
Здесь конъюгированна гипербилирубинемия носит более интересный характер.
У Дабина-Джонсона нарушен «отводящий насос» в апикальной части гепатоцита, через которую он в норме и должен выводиться. А у Ротора – нарушен обратный захват конюъюгированного билирубина белками OATP1B1 и OATP1B3 на базальной мембане, обращенной в кровоток. Т.е. конъюгированный билирубин по градиенту концентрации просачивается туда, куда не должен. Их генетических дефект приводит к тому, что контролирующие белки сбоят. Не сильно, но сбоят.
Синдром Ротора – это легкое подобие синдрома Дабина-Джонсона, болезнь мягкая, возникает в детском возрасте, нарушения активности печеночных ферментов нет ( а у Дабина может быть, хоть и небольшой, но подъем АЛТ/АСТ). Может быть интермиттирующая желтуха, но часто болезнь течет бессимптомно.
Основная причина синдрома Ротора – генетический дефект белков обратного захвата OATP1B1 и OATP1B3, непозволяющих в норме конъюгированный билирубину проходить через базальную мембрану в кровоток.
Синдром Люси-Дрисколл.
Транзиторная гипербилирубинемия новорождённых, связанная с присутствием у некоторых женщин в грудном молоке фактора, ингибирующего фермент УДФ-глюкуронилтрансферазу, что приводит к нарушению конъюгации билирубина. Отлучаем от груди – процесс конъюгации восстанавливается.
Синдром Байлера.
Является редкой сложной патологией, причиной которой служит мутация гена FIC1, кодирующего АТФазу Р-типа. Точная функция этого протеина неизвестна, но похоже, она связана с АТФ-зависимым транспортом фосфолипидов. Патология заключается в снижении канальцевого транспорта желчной кислоты, которое приводит к образованию аномальной желчи и постепенному разрушению канальцевой мембраны, что обусловливает возникновение холестаза. Но ГГТ при этом нормальный, так что истинного стаза там нет. Заканчивается все перипортальным фиброзом. Патология очень редкая. Манифестирует в в ПЕРВЫЕ НЕДЕЛИ жизни, носит семейный характер и характеризуется высокой гипербилирубинемией ( 300 мкмоль/л).
Синдром Аагенеса (Саммерскилла-Тигструппа, норвежский холестаз).
Проявляется нарушением функций печени вследствие гипоплазии ее лимфатических сосудов с развитием холестаза (семейный характер). Манифестация обычно наступает в неонатальном периоде с возможными рецидивами у взрослых. Хронический холестаз сопровождается дефицитом жирорастворимых витаминов. Дефицит витамина Е приводит к прогрессирующей мозжечково-спинальной дегенерации (синдром Пермуттера).
Синдром Мейленграхта.
Юношеская интермиттирующая гипербилирубинемия, относится к синдромам функциональной гипербилирубинемии. Происхождение скорее связано с ускоренным распадом эритроцитов, при этом данных в пользу гемолиза нет. Очень часто ее описывают как с-м Жильбера-2, практически неотличимы. Согласно данным литературы, в 90 % случаев заболевание манифестирует в 20—30-летнем возрасте. Намного чаще встречается у мужчин (10:1) по сравнению с женщинами. Болеют чаще работники умственного труда. В целом синдром Жильбера—Мейленграхта выявляется у 1—5 % населения.
Теория теорией, но о какой нозологии надо думать и с чем дифференцировать, когда пациент приносит анализ - а там оно. Что подозревать у пациента с гипербилирубинемией?
Свободная (неконъюгированная) гипербилирубинемия.
Чрезмерное образование (я не рассматриваю обильное подкожное кровоизлияние, оперативные вмешательства на печени)
1.1. Гемолиз
1.2. Болезнь Коновалова - Вилсона
Дефект захвата из кровотока гепатоцитами
2.1. Лекарственные препараты: противотуберкулезный рифамицин и многие противоопухолевые препараты
2.2. Синдром Жильбера (некоторые формы)
Дефекты конъюгации
3.1. Наследственные
• Синдром Жильбера (чаще всего)
• Синдром Криглера-Найяра 1 и 2 типов, если это ребенок
3.2. Приобретенные
• Хронические гепатиты ( прежде всего С)
• Цирроз в далекозашедшей стадии
• Гиперпродукция тироксина
• Неонатальные формы, включая от материнского молока
- Гемолиз (морфология эритроцитов, ретикулоциты и гаптоглобин)
- Б-нь Коновалова-Вильсона (кольцо Кайзера-Флейшера, церулоплазмин, медь в крови и моче)
- Диагностика гепатитов, цирроза (опрос, антитела и АСТ/АЛТ)
- ТТГ и Т3/Т4 Free на щитовидку.
Изолированная, конъюгированная гипербилирубинемия.
Мы сейчас будем говорить о редких вещах – когда конъюгированный билирубин повышен, а общая фракция – нет или повышена незначительно. При этом печеночные трансаминазы АСТ/АЛТ молчат и показатели холестаза в виде ЩФ и ГГТ тоже норма. При этом конъюгированный билирубин составляетоколо 50% от общего или превышает 7 мкмоль/л при повышении общего до 34,2 мкмоль/л. Это возможно только в 2 случаях:
- Дабин-Джонсона синдром ;
- Ротора синдром.
Обычно их не различают, поскольку тактика ведения/наблюдения не отличается в силу доброкачественного течения.
Свободная и конъюгированная гипербилирубинемия.
Такая сочетанная форма представляет наибольшие сложности и встречается наиболее часто. Важно составить диагностический алгоритм подхода. Самый главный вопрос начального этапа – гипербилирубинемия сочетается с измененными печеночными показателями ( АСТ-АЛТ-ГГТ-ЩФ) или нет.
Но это уже чуть позже, почти день потрачен на анализ данных ) И если читающие захотят…



